A Síndrome de Marfan é uma desordem genética do tecido conjuntivo, o qual fornece suporte estrutural e determina a elasticidade dos órgãos do corpo, ossos e ligamentos. Quem sofre da síndrome de Marfan apresenta estatura alta e os membros anormalmente longos (aracnodactilia, ou seja, as mãos e os pés têm um aspeto de patas de aranha).
Esta síndrome pode ainda atingir o tecido conjuntivo no coração, pulmões, olhos e ossos, bem como o sistema nervoso, obrigando os tecidos a esticar e a enfraquecer.
A condição foi descrita, pela primeira vez, pelo pediatra Antoine Marfan, em 1826, que lhe deu o nome, e pode afetar tanto homens como mulheres de qualquer etnia.
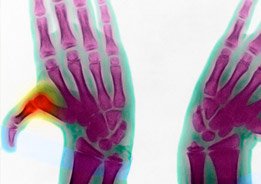 Imagem de raio-X das mãos de um doente com Síndrome de Marfan.
Imagem de raio-X das mãos de um doente com Síndrome de Marfan.Estima-se que atinja uma em cada cinco mil pessoas.
Causas da Síndrome de Marfan
A principal origem do problema é a existência de um único gene anormal (FBN1), que fica localizado no cromossoma 15, e que contém o código para fibrilina, uma proteína essencial para a formação das fibras elásticas do tecido conjuntivo. A fibrilina-1 e a fibrilina-2 são codificadas por dois genes distintos, o FBN1 e o FBN2, que ficam localizados nos cromossomas humanos 15 e 5. A mutação no FBN1 poderá causar a síndrome de Marfan.
Na maioria das vezes, este gene é herdado de um dos pais afetados. Cada vez que uma pessoa com síndrome de Marfan tem um filho, há 50% de hipóteses de a criança herdar a mutação da fibrilina e desenvolver a síndrome (herança autossómica dominante).
Em 25% dos casos, os pais podem não ser portadores desta desordem genética, mas pode acontecer devido a um espermatozoide ou óvulo afetado.
Sintomas da Síndrome de Marfan
Sabe-se que todas as pessoas que padecem deste problema possuem o gene FBN1. Contudo, os sintomas variam de doente para doente, mesmo dentro da própria família. A maioria nem irá manifestar sinais e desenvolver complicações da doença.
A acontecer, as anomalias manifestam-se ao nível de três sistemas principais:
- Esquelético – estatura elevada, escoliose, braços e mãos alongados e deformidade torácica.
- Cardiovascular – alterações da válvula mitral e dilatação da artéria da aorta. Os aneurismas da aorta e as dissecções da aorta são as complicações cardiovasculares mais graves, representando risco de morte.
- Ocular – provoca miopia e luxação do cristalino.
Tratamento da Síndrome de Marfan
A síndrome de Marfan é uma deformação congénita muito difícil de diagnosticar, uma vez que não existe nenhum teste específico para o efeito. Como se trata de uma pleiotropia (a mutação de um gene provoca alterações em vários órgãos), para identificar a doença, é necessário análise do historial clínico da família, recorrer a uma equipa médica e a exames multidisciplinares, como seja um ortopedista, geneticista, cardiologista e oftalmologista.
Embora não exista cura para a doença, a vigilância constante é fundamental, em especial durante a adolescência, uma vez que ajuda a:
- Controlar o crescimento esquelético e avaliar as mudanças na coluna e no esterno. As malformações esqueléticas podem ser passíveis de ser corrigidas por cirurgia.
- Monitorizar o tamanho e a função do coração através de eletrocardiogramas. Os doentes que têm síndrome de Marfan são, geralmente, medicados com betabloqueadores para diminuir a frequência cardíaca e o inotropismo (aumento da força da contração) do coração. Em caso de aneurisma da aorta, poderá ser preciso recorrer a cirurgia.
- Evitar problemas de visão. Na maioria dos casos, para corrigir defeitos nos olhos, utilizam-se óculos ou lentes de contato. A cirurgia pode ser necessária para algumas pessoas.
- Manter a boa higiene oral. Os doentes devem ter muito cuidado a este nível para evitar a entrada de bactérias e consequente endocardite.
Artigo revisto e validado pelo especialista em Medicina Geral e Familiar, José Ramos Osório.

Conteúdo revisto
pelo Conselho Científico da AdvanceCare.
Downloads
Consulte os nossos guias para hábitos saudáveis:
Sympton Checker
Utilize a nossa ferramenta de diagnóstico de sintomas.Programas AdvanceCare relacionados
Artigos relacionados
-
 Saúde e Medicina
Saúde e MedicinaCinco conselhos para um coração saudável
-
 Quotidiano
QuotidianoCuidar do sorriso é muito mais do que cuidar da boca
-
 Quotidiano
QuotidianoSaúde oral, saúde total