A retinopatia pigmentar (RP) constitui um grupo de doenças clínica e geneticamente heterogéneas, que têm em comum uma perda primária e progressiva dos fotorreceptores com alteração secundária das restantes camadas da retina. Nestas doenças ocorre tipicamente uma distrofia das células fotorrecetoras, nas quais um defeito genético causa a morte celular, principalmente dos bastonetes, e mais raramente dos cones e do epitélio pigmentado da retina (EPR).
A retinopatia pigmentar caracteriza-se pela perda progressiva da visão periférica, fraca visão noturna (nictalopia), ou deficiente visão com luz excessiva, podendo mais tarde causar perda da visão central. Inicialmente ocorrem alterações na visão noturna, mas com a evolução da doença, o campo visual tende a diminuir transformando-se apenas num ilhéu de visão central, normalmente chamada de visão tubular.
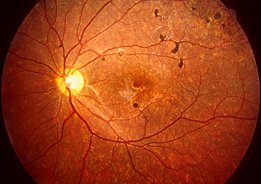 Degenerescência da retina (revelada pelos pigmentos visíveis).
Degenerescência da retina (revelada pelos pigmentos visíveis).A progressão e grau de perda visual variam de pessoa para pessoa.
Causas de Retinopatia Pigmentar
Há vários tipos de retinopatia pigmentar causados por diferentes mutações genéticas, que podem ser passadas de pais para filhos através de um dos três padrões de herança genética:
- Autossómica recessiva - os pais apresentam uma mutação genética no gene responsável pela doença, mas não apresentam sintomas, podendo ter filhos afetados ou não afetados.
- Autossómica dominante - um dos pais está afetado, mas pode ter ou não filhos afetados.
- Ligada ao cromossoma X – os homens são mais frequente e gravemente afetados. As mulheres, mesmo que apresentem uma mutação genética, apresentam perda de visão com menor frequência.
- Outras formas de hereditariedade estão também descritas (por exemplo, mitocondrial e dissomia uniparental).
A retinopatia pigmentar é tipicamente diagnosticada em adolescentes e adultos jovens, sendo que a idade de aparecimento e gravidade da patologia correlaciona-se com o tipo de hereditariedade:
- Hereditariedade ligada ao cromossoma X - 1ª década de vida.
- Hereditariedade autossómica recessiva – 2ª ou 3ª década de vida.
- Hereditariedade autossómica dominante – 2ª ou 3ª década de vida.
Sintomas de Retinopatia Pigmentar
Os sintomas dependem de como os bastonetes e cones são inicialmente afetados. Na maioria dos casos, os bastonetes são afetados primeiro, e estando eles mais concentrados nas porções periféricas da retina e sendo estimulados pela luz fraca, a sua degeneração afeta a visão periférica e noturna. Quando os cones, mais centralmente localizados, são afetados perde-se a perceção de cores e visão central. Os sintomas começam geralmente, entre os 10 e os 30 anos e a progressão da doença é variável, ocorrendo visão em túnel nas fases avançadas.
Os critérios de diagnóstico de retinopatia pigmentar são:
- Nictalopia (cegueira noturna).
- Perda progressiva da visão periférica.
- Disfunção dos bastonetes (dificuldade de adaptação escotópica e alteração primária do ERG escotópico).
- Perda progressiva da função dos fotorreceptores.
Tratamento de Retinopatia Pigmentar
O diagnóstico da patologia é realizado mediante exames adequados. Inicialmente existe preservação inicial da acuidade visual central com constrição progressiva dos campos visuais. Em fases avançadas, observa-se uma alteração da sensibilidade de contraste, da visão cromática e perda da visão central. O exame de biomicroscopia revela a existência de cataratas sub-capsular posterior precoce enquanto a fundoscopia permite identificar espículas pigmentadas periféricas, palidez do disco ótico, diminuição do calibre vascular e heterogeneidade do epitélio pigmentado da retina (EPR). Nas fases tardias, a mácula é também afetada com atrofia do EPR, edema macular cistoide e fibrogliose pré-retiniana.
Estão disponíveis testes genéticos para o diagnóstico da retinopatia pigmentar, que para além de permitirem avaliar o risco de transmissão da doença de pais para filhos, ajudam também a diagnosticar a doença. Isto revela-se uma mais-valia já que um doente adequadamente diagnosticado tem mais hipóteses de controlar a doença. Contudo, nem todos os genes que causam a doença foram descobertos, pelo que o resultado do diagnóstico genético pode não ser totalmente determinante.
Não há tratamento definitivo para a retinopatia pigmentar. A terapia nutricional com ácido de vitamina A e ácido docosahexaenóico (DHA) surgiu como um tratamento eficaz para muitos doentes. À semelhança das terapias genéticas, também as tecnologias para o desenvolvimento de agentes terapêuticos para os bastonetes têm vindo a progredir. Várias linhas de investigação estão em curso, incluindo ensaios clínicos com fármacos neuroprotetores, tecnologia com células encapsuladas (Neurotech) e terapia génica, tendo sido já implantados alguns microchips que melhoraram a função da retina.
Artigo revisto e validado pelo especialista em Medicina Geral e Familiar José Ramos Osório.

Conteúdo revisto
pelo Conselho Científico da AdvanceCare.
Downloads
Consulte os nossos guias para hábitos saudáveis:
Sympton Checker
Utilize a nossa ferramenta de diagnóstico de sintomas.Programas AdvanceCare relacionados
Artigos relacionados
-
 Quotidiano
QuotidianoCinco doenças oftalmológicas a que deve estar atento
-
 QuotidianoSaúde e Medicina
QuotidianoSaúde e MedicinaVisão e diabetes – qual a relação?
-
 Saúde e Medicina
Saúde e MedicinaRastreios: já fez o seu?